
মন্তু দেখলো ডাক্তার আপা টুনিকে ডেকে তার পরিবার, বাবা- মা, ভাইবোন নিয়ে গল্পে গল্পে এটা সেটা জিজ্ঞেস করছে। তখন মন্তু হুট করে জিজ্ঞেস করে বসলো, ” আপা, টুনি তো অনেকদিন হইলো তার বাপের বাড়ি যায়না, বাপ মায়ের অবস্থা শুইনা কি হইবো?”
ডাক্তার আপা : কারণ আছে মন্তু মিয়া। আসলে একটি শিশুর অবয়ব কেমন হবে, তার চারিত্রিক বৈশিষ্ট্য কেমন হবে, এগুলো নির্ভর করে তার বংশের যে জিন, তার ওপর। ঠিক অনুরূপভাবে তার রক্ত কেমন তৈরি হবে, সেটিও নির্ভর করে তার বাবা-মায়ের রক্তের কম্পোজিশন কেমন ছিল, বিশেষ করে হিমোগ্লোবিন তৈরির ক্ষেত্রে জেনেটিক যে কম্পোজিশন তার ওপর। সেখান থেকে একজোড়া জিন নিয়ে- বাবার থেকে একটি ও মায়ের থেকে একটি, দুটো জিন পেয়ে তার হিমোগ্লোবিন তৈরির প্রক্রিয়াটি সম্পন্ন হবে।
আর বংশগত রক্তসল্পতাজনিত একটি রোগ হলো Thalassemia. তাই কোনো বাবা কিংবা মায়ের রক্তে যদি থ্যালাসেমিয়ার জিন থাকে এবং সেখান থেকে বংশ পরম্পরায় এটি যদি সন্তানের মধ্যে সঞ্চারিত হয়, তাহলে কিন্তু ওই সন্তানেরও থ্যালাসেমিয়ার জিনের কারণে রক্তের হিমোগ্লোবিন তৈরির ক্ষেত্রে একটি বিপত্তি ঘটবে। যেমন বাবা মায়ের ছিল, ঠিক সেটা আসবে।
মন্তু : আপা, বাবা মায়ের যদি সেই সমস্যা থাকে তাইলে সেটা সন্তানের যে হইবো, তা কিভাবে হইবো?
ডাক্তার আপা: ওইতো জিনের কথা বললাম না? ওই জিনের নির্দিষ্ট কাজ আছে এখানে, বুঝিয়ে বলি তোমায়-
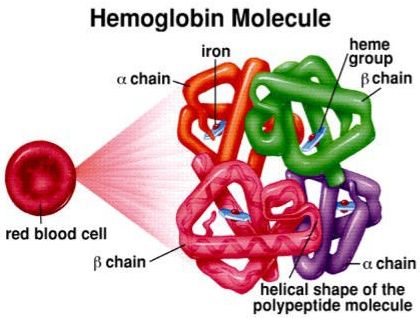
🔵 আমাদের রক্তের লাল অংশ যেটুকু তাতো এই Hb এর কারণে তাইনা? এখন, এই Hb এর দুইটি অংশ- একটি হলো heme আরেকটি হলো globin। এই globin তৈরির ক্ষেত্রে polypeptide chain থাকে। এক জোড়া Alpha chain আর এক জোড়া beta chain। এই দুই জোড়া চেইনের সমন্বয়ে গঠিত হয় globin। তার সঙ্গে heme মিলে হয় Hb। যাদের থ্যালাসেমিয়া রয়েছে, তারা হয় alpha chain অথবা beta chain একেবারেই তৈরি করতে পারে না কিংবা কম তৈরি করে। এতে তার Hb এর ঘাটতি হয়ে যাচ্ছে। যেটুকু তৈরি হচ্ছে সেটি ত্রুটিপূর্ণ।
ত্রুটিপূর্ণ হলো কেন? কারণ যে জিন দিয়ে globin chain টি তৈরি হচ্ছে, সেই globin যদি কম পরিমাণের হয় বা একটি chain যদি কম হয়, তাহলে সে replace করার জন্য কাছাকাছি যারা রয়েছে তার সঙ্গে সমন্বয় করবে। সমন্বয় হয়ে যে Hb তৈরি হয়, সেই Hb এর জীবন বেশি দিনের হয় না। এটি ১২০ দিনের আগেই ভেঙ্গে যাবে। এক্ষেত্রে রোগীদের RBC ১২০ দিনের পরিবর্তে ৩০/ ৪০ দিন বা ৫০ দিনেই ভেঙ্গে যাচ্ছে।

তাহলে একটি নেতিবাচক প্রভাব পড়বে। শিশু বয়সে bone marrow এর উৎপাদন ক্ষমতা কিছুটা বাড়তি থাকে- ৬ থেকে ১২ গুণ বেশি থাকে। এতে সে কিছু Hb কম তৈরি করলেও, bone marrow কিন্তু তখন বেশি বেশি কাজ করে। আস্তে আস্তে যখন বয়স বাড়তে থাকে, যারা বিশেষ করে Thalassemia minor বা carrier তাদের বয়স বাড়ার সঙ্গে সঙ্গে Hb তৈরির ক্ষমতা কমতে থাকে। একটি সময়ে এসে কিন্তু তাদের এনিমিয়া হয়।
পক্ষান্তরে যারা Thalassemia major বা intermediate তাদের যে Hb তৈরি হয় এটি আসলেই খুব কম এবং ত্রুটিপূর্ণ। ছোট বয়স থেকেই কিন্তু সে এনিমিয়ায় ভোগে।
মন্তু : আচ্ছা আপা, তাইলে এই থ্যালাসেমিয়া কি বিভিন্ন রকম হইবো?
ডাক্তার আপা : হ্যাঁ মন্তু, ঠিক ধরেছো। বিভিন্ন কিছুর ওপর ভিত্তি করে আমরা ভাগ করে থাকি। প্রথমেই তো দুইরকম থ্যালাসেমিয়া রয়েছে-
▪Beta-thalassemia ( Beta chain এর production কম হবে ফলে red cell এ Hb-A এর পরিমাণ কমে যাবে)
▪Alpha- thalassemia ( Alpha chain এর defective synthesise হবে ফলে Hb production কমে যাবে)
🔸 এখন এই Beta thalassemia কে আবার কয়েকটি ভাগে ভাগ করা হয়, যেমন-
🔹 On the basis of the extent of reduction of beta-chain synthesis :
➡ Beta+ thalassemia ( incomplete suppression of beta-chain synthesis)
➡ Beta০ thalassemia ( complete absence of beta-chain synthesis)
🔹 Clinical classification :
➡ Beta thalassemia major ( major or total suppression of beta-chain synthesis & homozygous form of the disease) এটা আবার দুইরকম –
▪Beta+ thalassemia (Hb-A is present in small amount)
▪Beta০ thalassemia (Hb- A is completely absent)
➡ Beta thalassemia minor (Suppression of beta chain synthesis is much less severe & is heterogeneous form of the disease)
➡ Thalassemia intermedia ( Severity of thise diseases lies between that of the major & minor forms)
🔸 Alpha thalassemia কেও কয়েকটি ভাগে ভাগ করা হয়, যেমন-
🔹 Silent carrier: 1 gene is deleted
🔹 Alpha- thalassemia trait: 2 genes are deleted
🔹 Hb-H disease: 3 genes are deleted
🔹 Hb-Bart’s hydrops fetalis: 4 genes are deleted
মন্তু : কি কি লক্ষণ দেখা যাইবো আপা এইডা হইলে?
ডাক্তার- আপা : হুম, এটার নির্দিষ্ট কিছু clinical findings রয়েছে। বলছি-
🔺 Symptoms :
▪Features of anemia
▪Jaundice ( mild to moderate)
▪Features of hypersplenism ( bleeding manifestations & infection)
🔺 Signs :
▪Clinical triad of anemia, jaundice, splenomegaly
▪Bony changes (Mongoloid facies)
▪Growth retardation
▪Complications ➡
✔ Infections
✔ Iron overload, caused by repeated transfusions damages the liver & endocrine organs. It also damages the heart & death occurs from CHF & cardiac arrythmias.
✔ The child fails to thrive, anorexia, diarrhoea, loss of body fat & recurrent fever occur.
✔ Osteoporosis
▪Occasionally epistaxis, skin pigmentation, leg ulcers, gall-stones.
মন্তু : এই এনিমিয়া হইলে কোন পরীক্ষা করতে হয়না আপা? কেমনে জানা যাইবো?
ডাক্তার আপা : হ্যাঁ মন্তু, আমরা বিভিন্ন পরীক্ষা- নিরীক্ষা করে দেখে থাকি। প্রথমত রক্ত পরীক্ষা করি, তখন blood picture দেখে কিছু জিনিস বুঝতে পারি-
🔹Hb% : Severely reduced (3-9 gm/dl) & RBC count : reduced
🔹MCV, MCH & MCHC all are reduced.
🔹Peripheral blood film-
➡ RBC :
▪Microcytic hypochromic cells with marked anisocytosis & poikilocytosis.
▪Some cells are macrocytic & there are occassional spherocytes.
▪Tear-drop cells are often seen. Target cells & basophilic stippling are prominent.
➡ Reticulocytes : Increased ( 10% or more)
➡ WBC : Leukocytosis
➡ Platelate : Normal
দ্বিতীয়ত আমরা দেহে Iron এর পরিমাণ পরীক্ষা করার জন্য biochemical tests করে কিছু জিনিস বুঝতে পারি-
🔹Serum bilirubin : Slightly increased
🔹Serum ferritin, iron,Total iron binding capacity, % saturation all are normal. কিন্তু repeated blood transfusion হলে এসব কিছু আবার বেড়ে যাবে।
তৃতীয়ত confirmatory test হিসেবে একটি test করা হয়, তা হলো-
🔹 Hb electrophoresis : Absence or almost complete absence of of Hb-A, with almost all the circulating Hb being Hb-F (10-98%). The Hb-A2 percentage is normal, low or slightly raised.
মন্তু : তাহলে এই থ্যালাসেমিয়া হইলে কি রকম চিকিৎসা দেওয়া হয় আপা?
ডাক্তার-আপা : এটা হলে কিছু treatment principal follow করা হয় মন্তু মিয়া, সেগুলো হলো-
✅ Regular blood transfusion
✅ General management ➡
▪ Avoid iron containing food
▪Folic acid supplementation
▪Zinc supplementation
✅ Splenectomy
✅ Bone marrow transplantation
✅ Induction of gamma-chain synthesis
✅ Gene therapy
(চলবে…)
Jinat Afroz Kiron (PMC, 2016-17)
Abhishek Karmaker Joy (SSMC, 2016-17)